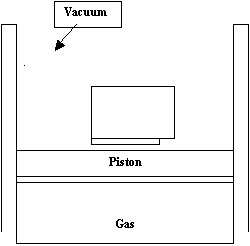
The Proper Definition of Pressure-Volume Work: Eric A. Gislason and Norman C. Craig Although thermodynamics is a mature discipline, some of its foundations are insecure. Two significantly different approaches to work and heat are in widespread use. Often the student is left in doubt about which approach is being used. We have been examining the two formulations of work and heat and the relationships between the two formulations. We now make a strong recommendation for using only one of the formulations [1-3]. There are two approaches in use to define pressure-volume work w. One, which the present authors strongly favor, is to define w (as well as the heat q) by making measurements in the surroundings before and after the process [1]. These definitions are referred to as “surr-based”. In general, the surroundings are made up of several parts, such as a piston, a calorimeter, the atmosphere, and an electrical system. The change in each part must be examined to determine w and q. Thus, a proper treatment of a process requires examination of the entire universe of the experiment (that is, the system plus all parts of the surroundings involved in the experiment) and leads naturally to the global formulation of thermodynamics [1,4,5], a powerful approach to thermodynamics. The alternative, system-based approach to determining w and q, which is also widely used, is to make measurements in the system before, during, and after the process to define both w and q [3]. These are referred to as “sys-based” definitions. More details for both approaches are given below. Here we simply note that authors using the surr-based approach [6] typically define pressure-volume work w as òPexdV, where Pex is some pressure external to the system, V is the volume of the system, and authors using the sys-based approach [7] define w as òPdV, where P is the system’s pressure. Let us now consider a specific example. An apparatus that will allow us to treat pressure and volume as independent variables is shown below:
The gas is the system. It is assumed that the piston can move up and down inside the containing cylinder, and there is a vacuum above the piston. There may be friction between the piston and cylinder. When the gas and piston are in mechanical equilibrium, the pressure exerted by the piston on the gas is given by mg/A, where m is the mass of the piston plus additional weights put on top of the piston, g is the acceleration due to gravity, and A is the cross sectional area of the piston. Thus, P = mg/A. We see that the equilibrium pressure P of the gas can be varied by varying m. Alternatively, we assume that the piston can be locked at any given height h above the bottom of the cylinder. In that case, the volume of the gas is given by V = hA, and the volume of the gas can be set to any given value by moving the piston up or down and then locking it into place. An alternative to the apparatus depicted in the figure is to have the atmosphere acting as the piston. This alternative description could apply to liquid and solid systems. Let us consider a process that involves the piston moving from a lower height h1 to a higher height h2. In what follows forces rather than pressures are initially used because frictional forces are difficult to picture as pressures. Nevertheless, the entire analysis can be recast in terms of pressures by dividing each force by the cross-sectional area A of the piston. There are three forces that act on the piston as it moves [1]. First, F is the instantaneous force exerted on the piston by the system, i.e., the gas. Note that F > 0, i.e., F is exerted upwards. By Newton’s third law, -F is the force exerted by the piston downwards on the system. If the system is at equilibrium, then F = PA, where P is the equilibrium pressure of the system. If, on the other hand, the piston is moving, the quantity F/A is often referred to as Ps, the instantaneous pressure exerted by the system on the surface of the piston. It must be emphasized that F and Ps are, in general, not measurable as the piston moves. The second force acting on the piston is –mg. The third force is Ffr, which includes all nonconservative (frictional) forces exerted on the moving piston by the surroundings. In the present case, at the very least, it would include any frictional forces between the piston and the cylinder. In all cases Ffr < 0 when the piston is moving up, and Ffr > 0 when the piston is moving down. The forces F and Ffr are normally not known unless the piston is at rest. Further discussion of these three forces is given in the earlier paper [1]. Now consider a process where the catch is removed, and the piston moves from height h1 to h2. We assume that the initial pressure of the gas exceeds mg/A, so the piston rises rapidly, overshoots h2, then falls, rises again, and oscillates until coming to rest at h2. In the general case the piston could have kinetic energy K1 when at h1 and K2 at h2, but we assume here for simplicity that K1 = K2 = 0. A well-known theorem of classical mechanics [1] states that the total work Wtot done on the piston by all of the forces during the process equals the net increase of kinetic energy of the piston. Wtot does not represent the thermodynamic pressure-volume work in either the surr-based or sys-based definition. For this experiment the theorem can be written
The total change in energy of the piston, which is purely mechanical, is given by DE(piston) = mg(h2 – h1). (2) This result for the piston involves only energy changes in the surroundings and is, in fact, the negative of w for this process obtained in the surroundings-based definition of w [1]. Thus, Eqs. (1) and (2) can be combined to give w(surr-based) = -DE(piston) =
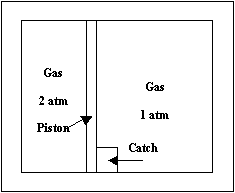
The last term on the right does not correspond to a traditional thermodynamic work term. Rather, it represents a conversion of some mechanical energy from the surroundings into thermal energy. This thermal energy is initially created in the surroundings but can end up in the surroundings or the system or partially in each. With surr-based definitions [1] it can be shown that the fraction of the thermal energy that ends up in the surroundings contributes to q, and the fraction that ends up in the system does not contribute to either q or w. By contrast, with sys-based definitions [3] the fraction of the thermal energy that ends up in the system contributes to q but not w, and the fraction that ends up in the surroundings does not contribute to either q or w. The system-based definition of pressure-volume work for the process considered here is [3] w(sys-based) = where the second integral uses the definition Ps = F/A discussed earlier. This definition is used by a large number of authors [7]. Equation (3) can now be rewritten as w(surr-based) = w(sys-based) It is apparent that surroundings-based and system-based definitions will not always give the same values of w, but they will when Ffr = 0. In a reversible process Ffr = 0 and P = Ps. Consequently, Eqs. (4) and (5) give w(sys-based, rev) = w(surr-based, rev) = The last term in Eq. (5) is positive, since Ffr has the opposite sign from dh whether the piston is moving up or down. Consequently, w(sys-based) £ w(surr-based) , (7) where the equality holds if and only if Ffr = 0, i.e., a frictionless piston. For most real processes Ffr ≠ 0, so system-based and surroundings-based work values will be different. An important exception is when the atmosphere acts as the piston and Ffr = 0. A first impression about Eq. (7) might be that one of the two definitions of w must be wrong, because the first law requires that DU = w + q. (8) In fact, the proper conclusions to draw are that the two definitions of q are different as well and that Eq. (8) is always valid. The surr-based and sys-based definitions of q are given in our recent papers [1,3]. Combining Eqs. (7) and (8) allows us to write for a given process that q(sys-based) ³ q(surr-based). (9) If the apparatus shown above is placed in a large water-bath calorimeter and has good thermal contact with it, then for the expansion process considered above [1] q(surr-based) = - CP(cal)DT = -DH(cal), (10) where CP(cal) and H(cal) are the constant-pressure heat capacity and enthalpy function of the calorimeter, and DT is the temperature change in the calorimeter. This definition is consistent because the calorimeter is in the surroundings. Note that q(surr-based) is given by the change in a state function of the calorimeter. From Eqs. (9) and (10) we immediately conclude that q(sys-based) cannot be related to a change in temperature of a calorimeter except in the special case where Ffr = 0. A more complete discussion of the two approaches to defining w and q are given in the author’s recent paper [3]. There we have fully developed the sys-based definitions and have given a number of reasons why surr-based definitions of work and heat are preferred. We highlight three here. First, in the definition of w(sys-based) in Eq. (4) the integral of Ps usually cannot be evaluated for irreversible processes, which means that w(sys-based) often cannot be determined. By contrast, w(surr-based) does not require the evaluation of that integral and w(surr-based) can be determined for irreversible processes. Second, w(surr-based) and q(surr-based) always require the evaluation of changes in thermodynamic state functions in the surroundings. In general, these quantities are straightforward to determine [see Eqs. (2), (3), and (10)]. By contrast, the integral ∫PsdV in Eq. (4) does not represent the change in any state property of the system. Third, we have shown [3] that w(sys-based) does not always satisfy the theorem of maximum work, which states that for a constant-temperature process –w ≤ -DA, where A is the Helmholtz free energy of the system. By contrast w(surr-based) always satisfies the theorem of maximum work. The superiority of surr-based definitions is illustrated by the following example. In 1964 Bauman [8] posed a simple thermodynamic process and challenged people to determine the work in the process. There was much discussion at the time [9-11] and one author [11] argued that the work could not be determined. In fact, we have determined the value for w [1], but it remains the only solution to date for this problem. The experiment is shown below. Two portions of an ideal gas at temperature T inside a closed cylinder of constant volume are separated by a massless, frictionless piston held by a catch. The piston has negligible heat capacity and can conduct thermal energy between the two subsystems. In addition, all surfaces of the rigid container are adiabatic so that the two samples of gas plus the piston are completely isolated from the rest of the universe. This arrangement guarantees that the two gases have the same final temperature T. There is 1 mol of gas on each side of the piston; one gas is initially at 2 atm and the other initially at 1 atm. When the catch is released, the piston initially moves toward the lower pressure gas but then oscillates back and forth with diminished oscillations around its final resting place. At the end each gas has a pressure of 4/3 atm and temperature T. A complete analysis is given in the earlier paper [1]. Here we simply note that if the high-pressure gas is the system, then w(surr-based) = -RT ln(4/3) and q(surr-based) = RT ln(4/3). In addition, DA = -RTln(3/2) for the high pressure gas, so –w + DA = RTln(8/9) < 0, as required by the theorem of maximum work. By comparison, ∫PsdV cannot be evaluated, so w(sys-based) and q(sys-based) cannot be determined. It is clear, then, why no other solution of this problem has appeared in the literature.
In conclusion, we emphasize two points about the development in this paper and other papers in this series [1-3]. First, work and heat can be defined using either surr-based or sys-based definitions, but the two sets of definitions often given different results. Nevertheless, the first law in the form of Eq. (8) is valid for both sets of definitions, and thermodynamics can be developed using either set of definitions. Since the definitions do give different results, it is critical that the definitions be clearly stated and used consistently. It is not acceptable to compute w from a surr-based definition and q from a sys-based definition or vice versa. Second, the authors strongly advocate using surr-based definitions [1] for work and heat. These definitions are more general and can be applied to irreversible processes where sys-based definitions cannot give either w or q. Handling real (that is, irreversible) processes is essential to a full realization of thermodynamics. References
Eric A. Gislason is Professor of Chemistry at the University of Illinois at Chicago. Norman C. Craig is Emeritus Professor of Chemistry at Oberlin College, Oberlin, OH 44074 |